Figure 15.3:
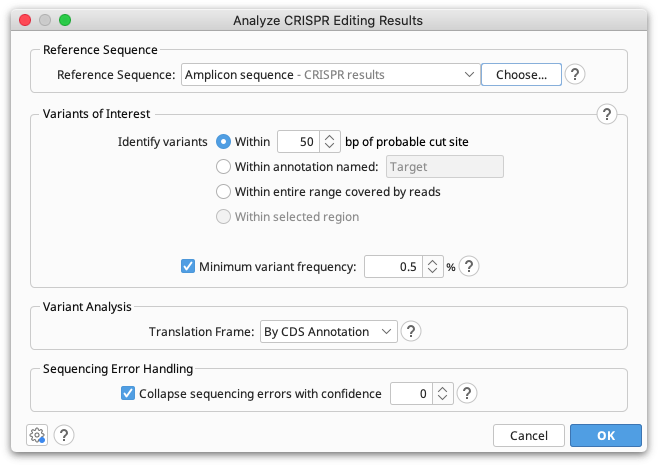
Analyze CRISPR editing results options dialog.
The reference sequence should be a short sequence spanning the CRISPR editing site, of similar length to the reads. This sequence is normally the unedited, or target sequence for calling variants against. The reference sequence can be selected together with the reads prior to opening the operation, or can be set from the operation dialog.
Workflows: The reference sequence option is not available from workflows. If this operation is included in a workflow, the reference sequence must be provided as input to the workflow. Or you can insert it into the workflow using the ’Add document chosen when running workflow’ option.
Only the portion of each read which spans the specified region of interest will be used for variant calling. This region can be either a specified number of bases around the probable cut site (default 50bp), the region currently selected in the sequence viewer, or the entire range covered by the reads.
Reads will be entirely excluded from variant calling if they match poorly on the ends of the reference sequence range matched by 99% of reads. See the algorithm overview for details.
The minimum variant frequency setting is used to exclude low frequency variants from the results displayed. Note that this setting does not change the reported frequencies of variants, i.e. the frequencies will be a percentage of both included and excluded variants.
The translation frame is used for calculating variant effects on the protein. The genetic code is obtained from the reference sequence properties which can be set in the Info tab or Sequence View.
Most of the time we can have reasonable confidence whether or not a rare variant is likely due to sequencing error and either correctly collapse it into the cluster it belongs to or correctly keep it separate. The setting Collapse sequencing errors with confidence controls what to do in borderline cases.
The value is log scale, so a value of +10 (or -10) means reads are collapsed (or not collapsed) with 90% confidence it is correct to do so, ±20 means 99% confidence, ±30 means 99.9% confidence.
Turning off this setting is equivalent to using a large positive value. For sequencing reads without Phred quality scores, each base is assumed to have quality score of 20 (99% confidence)