Figure 9.3:
The multiple alignment window
A multiple sequence alignment is a comparison of multiple related DNA or amino acid sequences. A multiple sequence alignment can be used for many purposes including inferring the presence of ancestral relationships between the sequences. It should be noted that protein sequences that are structurally very similar can be evolutionarily distant. This is referred to as distant homology. While handling protein sequences, it is important to be able to tell what a multiple sequence alignment means – both structurally and evolutionarily. It is not always possible to clearly identify structurally or evolutionarily homologous positions and create a single “correct” multiple sequence alignment (Durbin et al 1998).
Multiple sequence alignments can be done by hand but this requires expert knowledge of molecular sequence evolution and experience in the field. Hence the need for automatic multiple sequence alignments based on objective criteria. One way to score such an alignment would be to use a probabilistic model of sequence evolution and select the alignment that is most probable given the model of evolution. While this is an attractive option there are no efficient algorithms for doing this currently available. However a number of useful heuristic algorithms for multiple sequence alignment do exist.
The most popular and time-efficient method of multiple sequence alignment is progressive pairwise alignment. The idea is very simple. At each step, a pairwise alignment is performed. In the first step, two sequences are selected and aligned. The pairwise alignment is added to the mix and the two sequences are removed. In subsequent steps, one of three things can happen:
This process is repeated until a single alignment containing all of the sequences remains. Feng & Doolittle were the first to describe progressive pairwise alignment. Their algorithm used a guide tree to choose which pair of sequences/alignments to align at each step. Many variations of the progressive pairwise alignment algorithm exist, including the one used in the popular alignment software ClustalX.
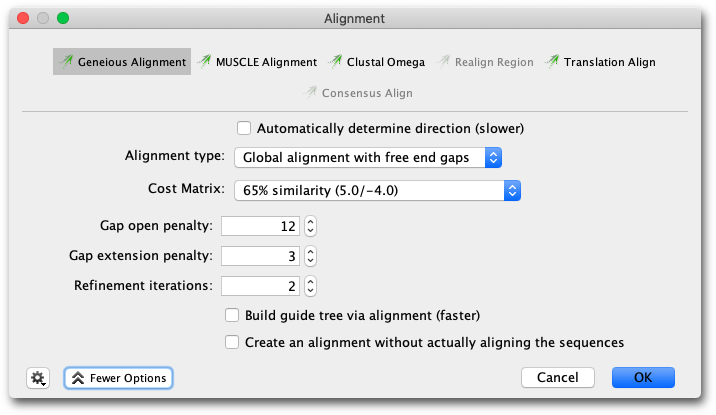
To run a multiple alignment using the Geneious aligner, select all the sequences you wish to align and click Align/Assemble →Multiple align.... Select Geneious as the alignment algorithm. The Geneious multiple alignment algorithm uses progressive pairwise alignment. The neighbor-joining method of tree building is used to create the guide tree.
As progressive pairwise alignment proceeds via a series of pairwise alignments, this function has all the standard pairwise alignment options, plus the option of refining the multiple sequence alignment once it is done. “Refining” an alignment involves removing sequences from the alignment one at a time, and then realigning the removed sequence to a “profile” of the remaining sequences. The number of times each sequence is re-aligned is determined by the refinement iterations option in the multiple alignment window. The resulting alignment is placed in the folder containing the original sequences.
A profile is a matrix of numbers representing the proportion of symbols (nucleotide or amino acid) at each position in an alignment. This can then be pairwise aligned to another sequence or alignment profile. When pairwise aligning profiles, mismatch costs are weighted proportional to the fraction of mismatching bases and gap introduction and gap extension costs are proportionally reduced at sites where the other profile contains some gaps.
In some cases building a guide tree can take a long time since it requires making a pairwise alignment between each pair of sequences. The build guide tree via alignment option may speed this part by taking a different route. First make a progressive multiple alignment using a random ordering, and use that alignment to build the guide tree. Notice that while this usually speeds up the process, it may not if the sequences are very distant genetically.
You can also do a multiple alignment via translation and back, as with pairwise alignment (see section 9.2.6 )