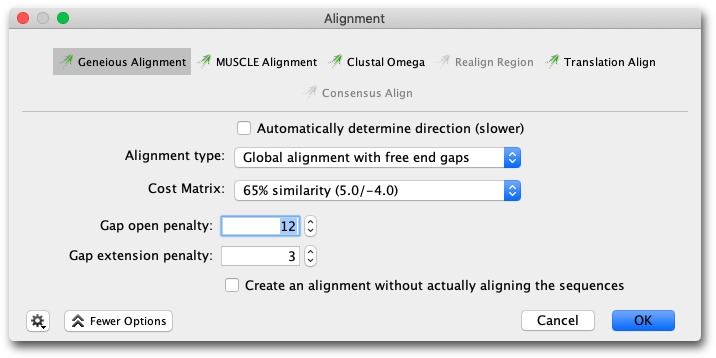
Figure 9.2:
Options for nucleotide pairwise alignment
There are two types of pairwise alignments: local and global alignments.
A local alignment is an alignment of two sub-regions of a pair of sequences. This type of alignment is appropriate when aligning two segments of genomic DNA that may have local regions of similarity embedded in a background of a non-homologous sequence.
A global alignment is a sequence alignment over the entire length of two or more nucleic acid or protein sequences. In a global alignment, the sequences are assumed to be homologous along their entire length.
In order to align a pair of sequences, a scoring system is required to score matches and mismatches. The scoring system can be as simple as “+1” for a match and “-1” for a mismatch between the pair of sequences at any given site of comparison. However substitutions, insertions and deletions occur at different rates over evolutionary time. This variation in rates is the result of a large number of factors, including the mutation process, genetic drift and natural selection. For protein sequences, the relative rates of different substitutions can be empirically determined by comparing a large number of related sequences. These empirical measurements can then form the basis of a scoring system for aligning subsequent sequences. Many scoring systems have been developed in this way. These matrices incorporate the evolutionary preferences for certain substitutions over other kinds of substitutions in the form of log-odd scores. Popular matrices used for protein alignments are BLOSUM and PAM1 matrices.
Note: The BLOSUM and PAM matrices are substitution matrices. The number of a BLOSUM matrix indicates the threshold (%) similarity between the sequences originally used to create the matrix. BLOSUM matrices with higher numbers are more suitable for aligning closely related sequences. For PAM, the lower numbered tables are for closely related sequences and higher numbered PAMs are for more distant groups.
When aligning protein sequences in Geneious, a number of BLOSUM and PAM matrices are available.
Once a scoring system has been chosen, we need an algorithm to find the optimal alignment of two sequences. This is done by inserting gaps in order to maximize the alignment score. If the sequences are related along their entire sequence, a global alignment is appropriate. However, if the relatedness of the sequences is unknown or they are expected to share only small regions of similarity, (such as a common domain) then a local alignment is more appropriate.
An efficient algorithm for global alignment was described by Needleman and Wunsch 1970, and their algorithms was later extended by Gotoh 1982 to model gaps more accurately. For local alignments, the Smith-Waterman algorithm is the most commonly used. See the references at the links provided for further information on these algorithms.
Like a dot plot, a pairwise alignment is comparison between two sequences with the aim of identifying which regions of two sequences are related by common ancestry and which regions of the sequences have been subjected to insertions, deletions, and substitutions.
To run a pairwise alignment using the Geneious aligner, select the two sequences you wish to align and choose Align/Assemble → Pairwise align.... The options available for the alignment cost matrix will depend on the kind of sequence.
The score of a pairwise alignment is:
matchCount × matchCost + mismatchCount × mismatchCost
For each gap of length n, a score of gapOpenPenalty + (n− 1) ×gapExtensionPenalty is subtracted from this.
Where
When doing a Global alignment with free end gaps, gaps at either end of the alignment are not penalized when determining the optimal alignment. This is especially useful if you are aligning sequence fragments that overlap slightly in their starting and ending positions, e.g. when using two slightly different primer pairs to extract related sequence fragments from different samples. You can also do a Local Alignment if you want to allow free end overlaps, rather than just free end gaps in one alignment.