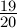 ∗ log(1 −
∗ log(1 −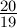 ∗ p)
∗ p)
The evolutionary distance between two amino acid sequences can be determined under the assumptions of a particular model of amino acid substitution. The substitution model defines a rate matrix that can be used to calculate the probability of evolving from one amino acid to another over a given time.
As with nucleotides, gaps are not penalized when using the Geneious Tree Builder. Sites with gaps are ignored when calculating pairwise distances (i.e., gaps are not treated as a 21st amino acid state).
This is the simplest substitution model. It assumes that all amino acids have the same equilibrium base frequency, i.e., each amino acid occurs with a frequency of 0.05 in protein sequences. This model also assumes that all amino acid substitutions occur at equal rates.
If the proportion of non-gap, non-ambiguous sites that are mismatched between the sequences is given as p, the formula for computing the distance between the sequences is:
d = − ∗ log(1 − ∗ p)
Under Jukes-Cantor the number of substitutions is assumed to be Poisson distributed with a rate of 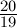 u, i.e., the probability of no substitutions at a given site over a branch of length ut is e−
u, i.e., the probability of no substitutions at a given site over a branch of length ut is e−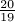 ut.
ut.
Technically, Jukes-Cantor for amino acid sequences is the Neyman model (Neyman 1971) with 20 states.