10.4.4 RNAseq mapping
To map RNA sequence reads to a genome with introns, choose Geneious RNA as the Mapper in the Map to Reference setup dialog. This function can map reads that span existing annotated introns, or discover insertions, novel introns and fusion genes.
This function works in the same way as deletion and structural variant discovery (section 10.4.3
) for DNA mapping, by analyzing how fragments of each read align to different regions of the reference sequence(s), and creating a junction annotation at the point where the read is split. By default, at least 2 reads must support the discovery of a junction in order for it to be annotated. This threshold can be adjusted under More Options by changing the Minimum support for intron/fusion gene discovery setting.
If Span annotated mRNA introns is checked, junctions will be created from existing annotations on the reference sequence. Reads are still allowed to map anywhere, but will be allowed to freely span these junctions if that produces the best mapping.
To only find introns up to a certain size, check Find novel introns up to...; to find introns of any size, insertions, or structural rearrangements that may indicate a fusion gene, use Find fusion genes and novel introns.
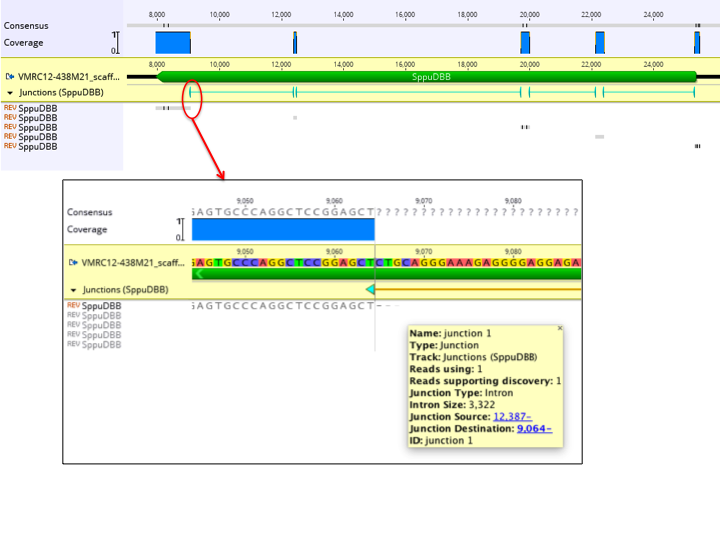
As for deletion and structural variant discovery, junctions are annotated on the reference sequence under a track named after the reads (see Figure 10.5
). Each junction has the following properties:
- Junction Type: This will be Insertion for short insertions. For introns under 2,000,000 bp, this will be Intron. For longer introns or structural variants, this will be Fusion, with (inversion) potentially appended.
- Intervals: Junctions of type Insertion are shown as one single interval annotation covering the gapped region in the contig, or when viewed on the unaligned reference sequence, positioned between the nucleotides on either side of the insertion. Junctions of type Intron and Fusion are each represented with two 1-bp annotation intervals positioned on the last nucleotide before the read jumps and continues elsewhere. For Introns, this is a single annotation with two linked intervals. Introns that have common start and finish nucleotides will be assigned an appropriate direction. For Fusions, the junction site is split into two separate annotations, each with a jagged edge on one side of the interval to indicate the side which jumps elsewhere.
- Intron Size: This is present when Junction Type is Intron.
- Fusion Distance: This is present when Junction Type is Fusion to indicate the distance between the two junction sites.
- Insertion Size: This is present when Junction Type is Insertion to indicate the number of nucleotides in the insertion.
- Insertion: This is present when Junction Type is Insertion to indicate the nucleotides inserted.
- Reads supporting discovery: Indicates the number of reads that supported discovery of this junction during the first pass. This may be lower than the advanced minimum support setting in cases where other reads supported discovery of a slightly offset version of this junction, which allows this junction to be retained on the next pass.
- Reads using: Indicates the number of reads that used this junction as part of their mapping during the second pass.
- Junction Source & Junction Destination: Clickable links to the junction positions in the reference sequence. When the destination is a different reference sequence, this is prefixed with the sequence name followed by a colon.
- Color: Annotations are colored from blue to green based on increasing values of Reads supporting discovery.
Reads spanning junctions may be represented in one of three possible ways.
- For insertions, the insertion is represented as a gap in the reference sequence.
- For introns under 15 bp, the deletion is represented as a gap in the read. This gap contributes towards calling a gap in the consensus sequence.
- For longer introns or for fusion genes, two copies of the read appear in the contig where the fragment of the read extending past the junction is marked as trimmed. Trimmed regions do not contribute to consensus sequence calling. These trim regions will only be visible when in editing mode. When not in editing mode, the trimmed regions will appear as 3 gaps fading to light grey. Clicking on these fading gaps will jump to the read at the other end of the junction. Faded gaps depend on the presence of the junction track created at the time of mapping. If these annotations are deleted, the faded gaps will appear as trimmed regions instead. (see Figure 10.5
).