The ![]() Statistics tab displays statistics about the sequence(s) being viewed. If only part of the sequence/alignment or assembly is selected then the statistics displayed will correspond to the highlighted part. The length of the sequence or part of the sequence is displayed next to the Statistics option.
Statistics tab displays statistics about the sequence(s) being viewed. If only part of the sequence/alignment or assembly is selected then the statistics displayed will correspond to the highlighted part. The length of the sequence or part of the sequence is displayed next to the Statistics option.
If the sequence is less than 14bp in length (Marmur and Doty 1962):
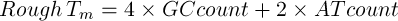
|
(5.1) |
If the sequence is greater than 13bp in length (Chester and Marshak 1993):

|
(5.2) |
A=71.0788 R=156.1875 N=114.1038 D=115.0886 C=103.1388 E=129.1155 Q=128.1307 G=57.0519 H=137.1411 I=113.1594 L=113.1594 K=128.1741 M=131.1926 F=147.1766 P=97.1167 S=87.0782 T=101.1051 W=186.2132 Y=163.1760 V=99.1326 U=150.0388 O=237.3018
For DNA sequences, the following values are used:
A=313.21 T=304.2 G=329.21 C=289.18
The DNA molecular weight assumes no modification of the terminal groups of the sequence.
If the sequence is a single-stranded, synthesised oligonucleotide (e.g. by primer extension), the value is adjusted for the removed phosphate group by using:
Molecular Weight = calculated molecular weight - 61.96
If the sequence is a single-stranded sequence cut by a restriction enzyme, the value is adjusted for the extra 5′-monophosphate left by most restriction enzymes by using:
Molecular Weight = calculated molecular weight - 61.96 + 79.0
For dsDNA, these values are adjusted for both strands.
For RNA sequences, the following values are used:
A=329.21 U=306.2 G=345.21 C=305.18
The RNA molecular weight assumes no modification of the terminal groups of the sequence. For a 5′-triphosphate group, weights are adjusted using
Molecular Weight = calculated molecular weight + 159.0
For both Identical sites and Pairwise % Identity, the statistics are calculated from the subset of sequences and nucleotides/amino acids selected. If just a single sequence is selected, the statistics are calculated as if all sequences are selected over the selected columns. The consensus sequence is always excluded from calculation of both of these values.
Selecting a sub-region of your contig will display statistics for just that region, including calculation of separate forward/reverse coverage on large contigs.
For contigs where reads extend outside the bounds of the reference sequence, the document table mean coverage is calculated excluding regions outside the reference sequence. The mean coverage displayed in the contig viewer statistics in this same situation when nothing is selected includes regions outside the reference sequence. Click on the name of the reference sequence to select just that region in order to display detailed coverage statistics over just the region spanned by the reference sequence.
For documents that are created or modified in Geneious 8.1 or later, the GC content can also be viewed in the %GC column in the document table.
The %GC column shows the percentage of A, C, G, T, U, S, W nucleotides that are either G, C, or S. Ambiguous bases that contain a mixture of GC and non-GC bases (e.g. R, Y, M, K) are excluded from the calculation. This field is available on all nucleotide sequences, contigs, alignments, and sequence lists that were created or had their sequences last modified in Geneious 8.1 or later. For contigs and alignments, the consensus sequence and reference sequence (if any) are excluded from the calculation.
For sequences within an alignment, contig or list, the %GC column only shows the overall value for the alignment. To see a table of GC percentages for all individual sequences within an alignment or contig, the sequences need to be extracted to stand-alone sequences. Alternatively, individual values can be viewed in the statistics panel by clicking on the name of the sequence to select it.
Sequences in a list or alignment can be sorted by GC content by right clicking in the sequence viewer and choosing Sort → %GC.