Figure 7.1:
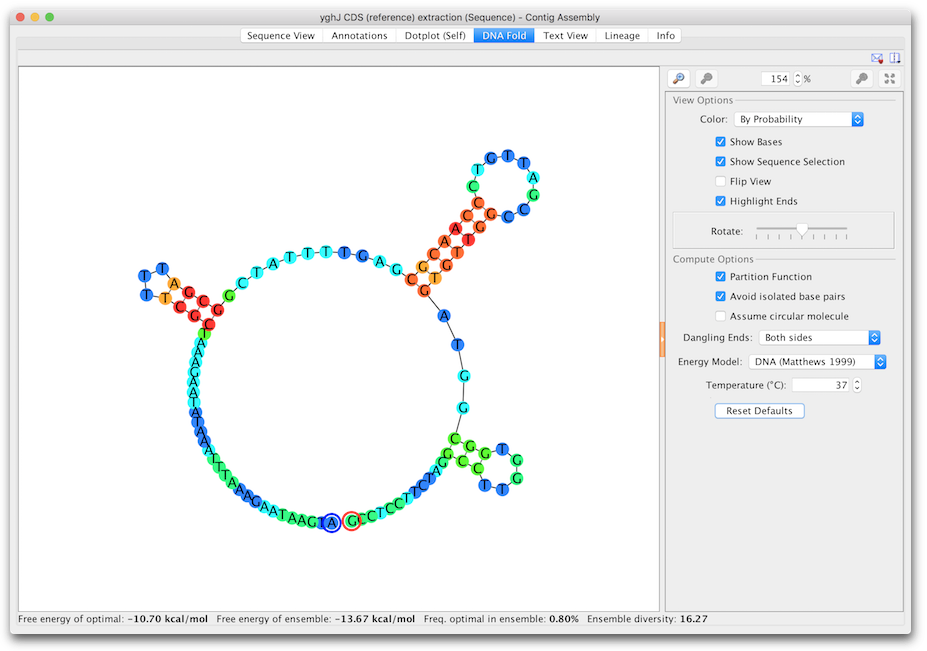
A view of an mRNA secondary structure prediction in Geneious Prime
By default this viewer is only shown when an oligo sequence is selected. If you wish to use RNA fold on a non-oligo sequence, go to Tools → Preferences → Appearance and Behavior and enable the option Show DNA/RNA fold view on all sequence. This will show the tab for any sequence less than 3000 bp. If the selected sequence is DNA, the tab will be labelled DNA Fold and if it is RNA it will be labelled RNA Fold (Figure 7.1 ).
The fold prediction is performed by the Vienna package RNAfold tool. Information on the options for this tool can be found at the following web page: http://www.tbi.univie.ac.at/~ivo/RNA/RNAfold.html.
The View Options allow you to turn off/on and color the bases, flip the coordinates, highlight the start (blue) and end (red) of the sequence and rotate the model. As with other viewers, you can zoom in on the model and drag the view around, or use the scrollwheel using the same keyboard modifiers as the sequence viewer. Selection is synchronized between the sequence view and the fold view. In addition, when in split view mode, the fold viewer will scroll to the selected area when zoomed in.
By default, Color by probability is used where red bases are the ones with the strongest probability of the bases being paired with each other in paired regions, or being unpaired in unpaired regions. Green is the middle ground and blue is the lowest probability. Color by probability is only available when using the Partition Function.
Compute Options will rerun RNAfold when you change their settings, so depending on the size of the sequence there may be a noticeable recompute time.